Artificial intelligence powers protein-folding predictions

Rarely does scientific software spark such sensational headlines. “One of biology’s biggest mysteries ‘largely solved’ by AI”, declared the BBC. Forbes called it “the most important achievement in AI — ever”. The buzz over the November 2020 debut of AlphaFold2, Google DeepMind’s artificial-intelligence (AI) system for predicting the 3D structure of proteins, has only intensified since the tool was made freely available in July.
The excitement relates to the software’s potential to solve one of biology’s thorniest problems — predicting the functional, folded structure of a protein molecule from its linear amino-acid sequence, right down to the position of each atom in 3D space. The underlying physicochemical rules for how proteins form their 3D structures remain too complicated for humans to parse, so this ‘protein-folding problem’ has remained unsolved for decades.
Researchers have worked out the structures of around 160,000 proteins from all kingdoms of life. They have been using experimental techniques, such as X-ray crystallography and cryo-electron microscopy (cryo-EM), and then depositing their 3D information in the Protein Data Bank. Computational biologists have made steady gains in developing software that complements these methods, and have correctly predicted the 3D shapes of some molecules from well-studied protein families.
Despite these advances, researchers still lacked structural information for around 4,800 human proteins. But AlphaFold2 has taken structure-prediction strategies to the next level. For instance, an independent analysis by researchers in Spain showed1 that the algorithm’s predictions had reduced the number of human proteins for which no structural data was available to just 29.
“Based on CASP14 [results], they could get about two-thirds of the proteins with experimental accuracy overall, and even for hard targets, they can fold about one-third of the proteins with experimental accuracy,” says Yang Zhang, a biological chemist at the University of Michigan in Ann Arbor, whose algorithm was among CASP14’s runners-up. “That’s a very amazing result.” Two subsequent Nature papers2,3 and dozens of preprints have further demonstrated AlphaFold2’s predictive power.
Zhang considers AlphaFold2 to be a striking demonstration of the power of deep learning, but only a partial solution to the protein-folding problem. The algorithm can deliver highly accurate results for many proteins — and some multi-protein complexes — even in the absence of structural information. This could drastically accelerate experimental structural biology and help to guide research in protein engineering and drug discovery.
But many essential details remain out of reach for some proteins. Chris Sander, a computational biologist at the Dana-Farber Cancer Institute in Boston, Massachusetts, notes that algorithms still struggle with complicated protein targets that have multiple functional domains or highly dynamic structures. “It’s great what they’ve done,” says Sander. “But the flexibility of proteins and how they change is not touched by that, and just having a single snapshot doesn’t solve the problem of biological function.”
Progress in deep learning — and a growing community of AlphaFold2 users — could bring some of these challenges to heel, but a comprehensive understanding of protein biology will require a much broader computational and experimental toolbox.
Higher education
Deep learning incorporates machine-learning strategies in which computational neural networks are trained to recognize and interpret patterns in data. “These models don’t try to predict the structure all in one go,” says David Baker, a computational biologist at the University of Washington in Seattle. “They’re more like a physical simulation where the models are learning how to make good moves to improve the structure.” By training these algorithms with vast amounts of annotated experimental data, they can begin identifying links between sequence and structure that inform predictions for new proteins.
Over the past five years, multiple teams have made headway in applying deep learning to structure prediction. The first iteration of AlphaFold won CASP13 in 2018, but its performance was nowhere near the stand-out victory seen last year. Several academic laboratories subsequently developed deep-learning-based algorithms that outperformed the first generation of AlphaFold, including the Zhang lab’s D-I-TASSER4, the Baker lab’s trRosetta5 and RaptorX6, developed by Jinbo Xu and his team at the Toyota Technological Institute in Chicago, Illinois.
But these algorithms were generally applied as parts of a larger software pipeline, creating the potential for error and inefficiency. “You often had different components miscommunicating or not communicating optimally with one another because they were built piecemeal,” says Mohammed AlQuraishi, a systems biologist at Columbia University in New York City. These limitations have fuelled interest in end-to-end algorithms that manage the entire process from sequence to structure. DeepMind senior research scientist John Jumper, who is based in London, says that after CASP13, his team essentially discarded the first-generation AlphaFold and began to develop such a solution — AlphaFold2.
Several aspects of AlphaFold2 build on established techniques. For example, the algorithm begins by generating multi-sequence alignments (MSAs), in which a new protein with unknown structure is compared against related sequences from other species. By identifying co-evolving amino acids that change in parallel, algorithms can home in on those that are most likely to associate with each other in the folded protein — places where one change in the sequence requires compensatory mutations to preserve the overall structure.
Sander and his collaborator, computational biologist Debora Marks at Harvard University in Cambridge, Massachusetts, and their team developed this co-evolution-based technique in 20117. “It was the first solution that worked across the board for many proteins, using evolution to get the correct fold and the basic shape,” says Sander. “And now machine learning makes it even better.”
AlphaFold2’s developers drew on an unprecedented amount of information to build their MSAs, using billions of protein sequences from a data set compiled by computational biologist Martin Steinegger at Seoul National University in South Korea and Johannes Söding at the Max Planck Institute for Biophysical Chemistry in Göttingen, Germany. “They wanted me to turn that into a searchable database,” Steinegger says.
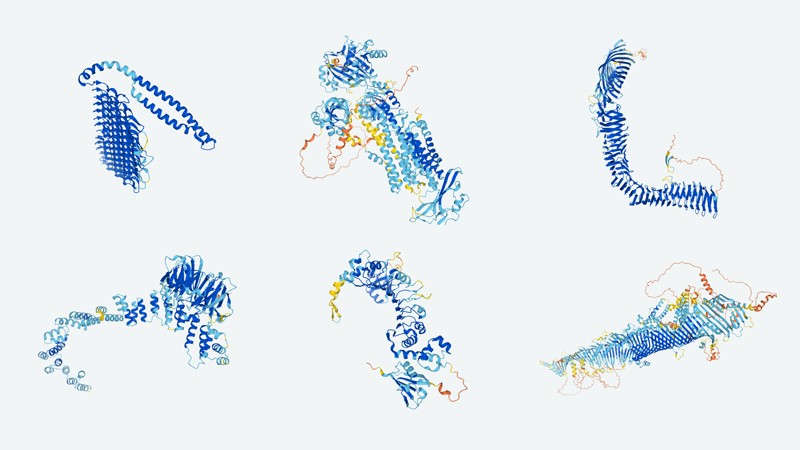
These predictions generated by AlphaFold2 highlight the structural variety of proteins.Credit: DeepMind
The DeepMind team also devised innovative solutions to the protein-folding problem. One is the use of pattern-recognition tools known as transformers, which are commonly used in image analysis and natural-language processing. Transformers are designed to recognize local patterns — strings of words or adjacent visual elements, for instance — that might guide interpretation of the data. DeepMind adapted them to work in the more challenging terrain of protein structure, building transformers that identify and focus on long-range protein interactions that are likely to be important in the final folded form. “In the final protein structure, you’ll make connections between quite distant things — like maybe residue 10 will talk to residue 350,” says Jumper.
The AlphaFold2 process simultaneously tackles protein folding from multiple angles, and generates multiple representations of the predicted structure in parallel. These are then compared, and the resulting insights help to refine the modelling process in subsequent iterations. Jumper and his colleagues enabled this by designing a neural-network architecture that allows fluid and efficient information exchange between components of the software. “I think the biggest thing that made this what it is was that very well-engineered communication system,” says AlQuraishi.
Prediction for the people
Because of the lag between AlphaFold2’s debut and the papers being published, and uncertainty among academics over whether full details would be made available, Baker and his postdoc Minkyung Baek worked from sparse information on the software’s architecture to develop their own version, RoseTTAFold8. This uses many of the same strategies as AlphaFold2, but with a few distinctive twists.
Since AlphaFold2’s July release2, labs have clamoured to work with the software and its structure predictions, which are available through a database hosted by the European Bioinformatics Institute.
Users generally find the software straightforward to use, although they need several terabytes of disk space to download the databases and multiple graphic processing units (GPUs) to handle the analysis. “Single-structure computations are not that bad — we run it for a couple of hours,” says bioinformatician Arne Elofsson at Stockholm University. But because of their scale and the resources required, analyses of the full complement of an organism’s proteins, or proteome, are likely to be out of reach for most academic labs for the time being.
For researchers who wish to test-drive the software, Steinegger and his colleagues developed ColabFold, a cloud-based system that runs both AlphaFold2 and RoseTTAFold using remote databases and computing power provided by Google9. The web-based interface is relatively simple: “You can plug in your sequence and then just push a button and it predicts the structure for you,” says Steinegger. But it also allows users to tinker with settings and optimize their experiments — such as by changing the number of iterations of structure prediction.
Finding the fold
Even the DeepMind team was taken aback by how well AlphaFold2 performed at CASP14. “We obviously had internal benchmarking that suggested that we were going to do very well,” says Jumper. “But at the end of the day, there was still a feeling in the back of my mind: is this really, really true?”
In August, researchers led by bioinformatician Alfonso Valencia at the Barcelona Supercomputing Center in Spain independently concluded1 that AlphaFold2 boosted the proportion of amino acids in human proteins that can be accurately mapped from 31% to 50%.
Zhang expects the software will make short work of the proteome’s low-hanging fruit. “They can probably fold all the single-domain proteins,” he says. But many proteins remain a challenge, such as those comprising multiple, independent, functional units joined by relatively flexible linker elements. In these cases, individual domains might fall in line, but their orientation relative to one another might not.
Even more challenging are protein segments that are intrinsically disordered in their natural state, which could represent more than one-third of all amino acids in the human proteome3. No algorithm can currently predict how these fold, but Jumper notes that extremely low pLDDT scores can at least demarcate these segments in a structure. “A totally unconfident prediction is quite a strong indicator of disorder,” he says.
One unexpected feature of both AlphaFold2 and RoseTTAFold is their ability to predict accurate structures from pairs of protein chains that form complexes called homodimers (if formed of two identical proteins) or heterodimers (formed of two different proteins) — something they were not initially trained to do.
Elofsson and his team have reported that they successfully modelled up to 59% of the two-protein complexes10 that they analysed using AlphaFold2. This process becomes more computationally challenging when attempting to identify likely complexes from scratch than when modelling known interacting pairs. But Baker and his team showed11 that, by applying multiple deep-learning algorithms in tandem, they were able to both identify and model hundreds of multi-protein complexes from millions of possible interacting pairs in the proteome of the yeast Saccharomyces cerevisiae. “RoseTTAFold was about 100 times faster [than AlphaFold2], and so we could run it on all pairs and then use it to filter out the ones that were most likely interacting,” says Baker. “Then we ran AlphaFold2 on that much smaller subset.”
Sensing the enthusiasm for this application, in October, DeepMind released AlphaFold-Multimer, which is specifically trained to tackle complexes of proteins that are formed by assemblies of multiple chains12. AlphaFold-Multimer generated high-accuracy predictions of interactions for 34% of the homodimeric complexes tested, and for 23% of heterodimeric complexes.
Functional frontiers
Still, many questions remain out of reach, notes Marks. “If your technology is bent on really learning to copy crystallography very well, then that’s great,” she says. But such static structural snapshots will not be suitable for exploring questions that relate to the manipulation or inherent dynamic behaviour of a given protein, she points out.
AlphaFold2 is also generally not suitable for predicting how individual amino acid changes alter protein structure — a crucial factor in understanding how mutations contribute to disease. This is in part because the algorithm uses evolutionary perspectives to converge on a correct solution from many slightly different sequences, says Stein, whose work focuses on characterizing such variants. “If you flip a single residue somewhere, you can’t expect it to suddenly say, ‘this is a disaster’,” she says. However, she and her team have found that they can couple wild-type protein structures generated by deep learning with other mutation-analysis algorithms to achieve more-accurate predictions13.
The good news is that structural biologists won’t be out of a job any time soon. In fact, they might now be able to devote more time to other pressing questions in the field. Structural biologist Randy Read at the University of Cambridge, UK, notes, for example, that structure predictions from AlphaFold2 are already helping crystallographers to drastically accelerate their data interpretation by overcoming the tedious ‘phase problem’ — a challenge associated with the interpretation of incomplete data generated in an X-ray diffraction experiment.
Protein designers could also see benefits. Starting from scratch — called de novo protein design — involves models that are generated computationally but tested in the lab. “Now you can just immediately use AlphaFold2 to fold it,” says Zhang. These results can even be used to retrain the design algorithms to produce more-accurate results in future experiments.



Post a Comment